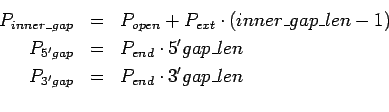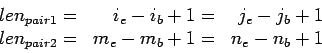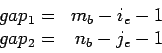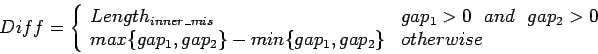

Up: LTR_FINDER Home Page
LTR_FINDER USER MANUAL
version 1.0.2
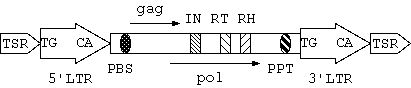
Figure 1: basic structure of a full-length LTR retrotransposon
The typical structure of a full-length LTR retrotransposon is shown
in Figure 1:
- LTR Region: 5'LTR and 3'LTR are two similar regions.
They are identical while the element inserts into the host genome,
and once inserted, they begin to evolve independently. Mutations and
indels thus are often found. A typical LTR retrotransposon has a
structure called TG..CA box, with TG at the 5' extremity of 5'LTR
and CA at the 3' extremity of 3'LTR.
- TSR Region: TSR(target site repeat) is a 4
~6bp short
direct repeat string flanking the 5' and 3' extremities of an
element. It is the sign of insertion of transposable elements.
- PBS: Near 3' end of the 5'LTR, there is a
~18bp
sequence complemented to the 3' tail of some tRNA. The site is very
important because tRNA binding process is the first step of
initiating reverse transcription.
- PPT: Polypurine tract is a short rich purine segment,
about 11
~15 bp in length. Like PBS, this region is important
for reverse transcription.
- Protein domains: In a typical virus genome there are three
polygenes: gag, pol and env. Among them, pol is most conserved.
Inside pol there are three important domains: IN(integrase),
RT(reverse transcriptase) and RH(RNase H), which are enzymes for
reverse transcription and insertion. RT and IN are regarded
essential for autonomous LTR elements to fulfill their function.
- These signals may become blur or even undetectable for evolutionary events.
The Program first constructs all exact match pairs by a suffix-array based algorithm and extends them to long highly similar pairs. Then Smith-Waterman algorithm is used to adjust the ends of LTR pair candidates to get alignment boundaries. These boundaries are subject to re-adjustment using supporting information of TG..CA box and TSRs and reliable LTRs are selected. Next, LTR_FINDER tries to identify PBS, PPT and RT inside LTR pairs by build-in aligning and counting modules. RT identification includes a dynamic programming to process frame shift. For other protein domains, LTR_FINDER calls ps_scan (from PROSITE, http://www.expasy.org/prosite/) to locate cores of important enzymes if they occur. Then possible ORFs are constructed based on that. At last, the program reports possible LTR retrotransposon models in different confidence levels according to how many signals and domains they hit.
LTR_FINDER accepts only FASTA format sequences, and only the first
ungapped string(identifier) in the description line is recorded to
identify the input sequence, other options in the description line
will be ignored. Here is an example of input:
>CHR1 19971009 Chromosome I Sequence
CCACACCACACCCACACACCCACACACCACACCACACACCACACCACACCCACACACACA
CATCCTAACACTACCCTAACACAGCCCTAATCTAACCCTGGCCAACCTGTCTCTCAACTT
ACCCTCCATTACCCTGCCTCCACTCGTTACCCTGTCCCATTCAACCATACCACTCCGAAC
... ... ... ...
TGATGGAGAGGGAGGGTAGTTGACATGGAGTTAGAATTGGGTCAGTGTTAGTGTTAGTGT
TAGTATTAGGGTGTGGTGTGTGGGTGTGGTGTGGGTGTGGGTGTGGGTGTGGGTGTGGGT
GTGGGTGTGGTGTGGTGTGTGGGTGTGGTGTGGGTGTGGTGTGTGTGGG
>CHR2 19970727 Chromosome II Sequence
AAATAGCCCTCATGTACGTCTCCTCCAAGCCCTGTTGTCTCTTACCCGGATGTTCAACCA
AAAGCTACTTACTACCTTTATTTTATGTTTACTTTTTATAGGTTGTCTTTTTATCCCACT
TCTTCGCACTTGTCTCTCGCTACTGCCGTGCAACAAACACTAAATCAAAACAATGAAATA
... ... ... ...
Users could submit sequences large to 50,000,000 bytes. The timeout limit for uploading sequence is 60 minutes. For users who want to scan very large size sequences, executive binary code will be available on request.
LTR_FINDER offers three types of output: Full output, Summary output and Figure output.
An example of Full output format is presented as follows:
>Sequence: CHR2 Len:813138
[1] CHR2 Len:813138
Location : 29632 - 35590 Len: 5959 Strand:+
Score : 9 [LTR match score:1]
Status : 11111111100
5'-LTR : 29632 - 29963 Len: 332
3'-LTR : 35259 - 35590 Len: 332
5'-TG : TG , TG
3'-CA : CA , CA
TSR : 29627 - 29631 , 35591 - 35595 [ATAAT]
Sharpness: 0.479,0.52
Strand + :
PBS : [17/22] 30031 - 30052 (ThrAGT)
PPT : [11/15] 35215 - 35229
Domain: 31889 - 32416 [possible ORF:31193-35236, (IN (core))]
Domain: 33779 - 34387 [possible ORF:31193-35236, (RT)]
Details of exact match pairs:
35259-35472[214] (1) 35474-35590[117]
29632-29845[214] (1) 29847-29963[117]
Details of the LTR alignment(5'-end):
|35259
CATTAGATCTATTACATTATGGGTGGTATGTTGGAATAAAAATCAACTATCATCTACTAAC
|| || | ||| ||| | |||*|||||||||||||||||||||||||||||
CA--AG--C----ACA-TAT-AAT----TGTTGGAATAAAAATCAACTATCATCTACTAAC
**-***----|29632
Details of the LTR alignment(3'-end):
35590|*****
ACAATTACATCAAAATCCACATTCTCTACAATAATAGAA--TAATGAA-CGATAACACACA
|||||||||||||||||||||||||||||* | ||| || || || | ||
ACAATTACATCAAAATCCACATTCTCTACA-TGGTAGCGCCTA-TGCTTCGGTTACTT---
29963|
Details of the PBS alignment(+):
tRNA type: ThrAGT
GCTTCCAAT----CGG-ATTTG
||||||||| ||| |||||
GCTTCCAATTTACCGGAATTTG
|30031
Details of PPT(+):
AACAAACAAATGGAT
|35215
While most of the output is straight forward, there are some fields
need further explanation.
- Score
Score is an integer varying from 0 to 11. It measures if signals
(TSR, TG..CA box, PBS, PPT, IN(core), IN(c-term), RT, RH) occur.
Because TG..CA box consists of four parts: TG at 5' end of 5'LTR, CA
at 3' end of 5'LTR, TG at 5' end of 3'LTR and CA at 3' end of 3'LTR,
there are 11 signals in total. The LTR match score (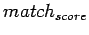 )
is the sequence similarity between 5'LTR and 3'LTR. It is a decimal
between 0 and 1.
)
is the sequence similarity between 5'LTR and 3'LTR. It is a decimal
between 0 and 1.
- Status
Status is an 11 bits binary string, each bit indicates the status of
a certain signal. From left to right, signals are: TG in 5' end of
5'LTR, CA in 3' end of 5'LTR, TG in 5' end of 3'LTR, CA in 3' end of
3'LTR, TSR, PBS, PPT, RT, IN(core), IN(c-term) and RH. If a signal
occurs, corresponding position is 1 and 0 otherwise.
- Sharpness
It is a decimal between 0 and 1 to evaluate the fineness of the
boundary of LTR region. Higher sharpness means more accurate
boundary decision. The first value is sharpness of 5' end and the
second is that of 3' end. In a window of length 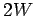 , Sharpness of
the center position is:
, Sharpness of
the center position is:
where 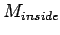 and (
and (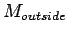 ) are the number of matched
bases in left half and right half window respectively. Put the
center position at the LTR boundary to get the sharpness.
) are the number of matched
bases in left half and right half window respectively. Put the
center position at the LTR boundary to get the sharpness.
- PBS & PPT
For PBS, the first number in square brackets is number of matched
bases and the second is total alignment length. For PPT, the first
is the number of purines and length of putative PPT. Following is
signal positions. String in parentheses is the tRNA type and
anti-codon(See this web page for detail:
http://lowelab.ucsc.edu/GtRNAdb/legend.html). The minus
sign before tRNA type stands for reverse strand(not showed in this
example).
- Details of exact match pairs
This section shows the exact math pairs used to construct the LTR
alignment. Number in square brackets is the pair length and number
in parentheses is the distance between neighboring exact match
pairs.
- Details of the LTR alignment
This section shows the alignment details around 5' and 3' boundaries
of LTR regions. Single asterisk in `|' line points out
putative boundary after the second run boundary decision (see
Strategy Section). Other 4~6 continuous asterisks show the
positions of putative TSR.
- Details of PBS & PPT
Numbers indicates the 5' ends of signals.
Summary output is extracted from Full-output by omitting some detailed information.
Here is an example output:
>Sequence: CHR2 Len:813138
[1] CHR2 Len:813138
Location : 29632 - 35590 Len: 5959 Strand:+
Score : 9 [LTR match score:1]
Status : 11111111100
5'-LTR : 29632 - 29963 Len: 332
3'-LTR : 35259 - 35590 Len: 332
5'-TG : TG , TG
3'-CA : CA , CA
TSR : 29627 - 29631 , 35591 - 35595 [ATAAT]
Sharpness: 0.479,0.52
Strand + :
PBS : [17/22] 30031 - 30052 (ThrAGT)
PPT : [11/15] 35215 - 35229
Domain: 31889 - 32416 [possible ORF:31193-35236, (IN (core))]
Domain: 33779 - 34387 [possible ORF:31193-35236, (RT)]
If user select output with figure, LTR_FINDER will produce a PNG file to show the relative position of each LTR retrotransposons. The figure was plotted by both normal axis and logarithmic axis. Elements drawn on silver background was plotted by their real size, that means, 1 pixel stand for 1 base. Elements drawn on white background was plotted under logarithmic axis, so that the long distance could be resized to place on a small canvas. Blue circles denote for PBS, brown circles stand for PPT, and purple circles on the end of LTRs are TSRs.
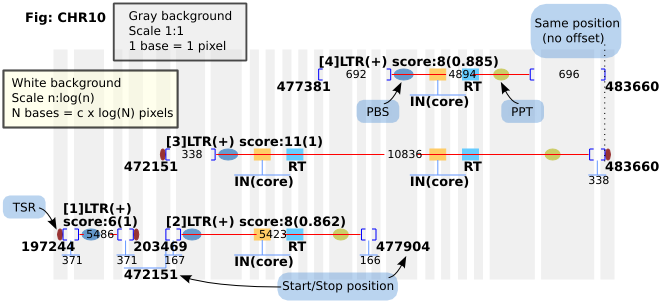
Figure 2: Legend of output figure
LTR_FINDER has many parameters, which can be divided into two
groups: parameters used in finding LTR retrotransposons
(construction parameters) and parameters used in filtering
out unreliable results (filter parameters). The first group
includes -o, -t, -e, -m,
-u, -D, -d, -L, -l,
-p, -g, -J, -j, -s,
-a and -r; the second group includes -S,
-B, -b, -w, -O, -P and
-F.
The five parameters control alignment algorithm. Denote gap open
penalty as 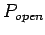 , gap extend penalty as
, gap extend penalty as 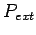 , gap end
penalty as
, gap end
penalty as 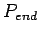 , match score as
, match score as  and mismatch score
as
and mismatch score
as 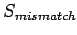 , then global and local alignments score are :
, then global and local alignments score are :
where 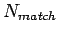 is the number of match bases;
is the number of match bases; 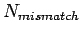 the number of unmatched bases; and
the number of unmatched bases; and
where
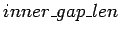 is the length of gap minus 2 (end bases).
Usually, is greater than .
is the length of gap minus 2 (end bases).
Usually, is greater than .
Distance between LTRs is:
The four parameters makes detected models meet common features of
LTR retrotransposons.
The three parameters control the extension of LTRs. An example of 2
neighbouring LTR pairs is shown in Figure 3. If
![$s[i_b\ldots m_e]$](img18.gif) and
and
![$s[j_b\ldots n_e]$](img19.gif) are similar enough, we extend LTRs from
are similar enough, we extend LTRs from
![$s[i_b\ldots i_e]$](img20.gif) and
and
![$s[m_b\ldots m_e]$](img21.gif) to
and
.
to
and
.
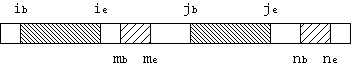
Figure 3:
![$P_1\{s[i_b\ldots i_e], s[j_b\ldots j_e]\}\ and \ P_2\{s[m_b\ldots m_e], s[n_b\ldots n_e]\}$](img22.gif)
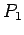 and
and 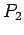 are pre-sorted so that
are pre-sorted so that 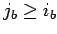 ,
, 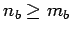 and
and 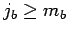 . Since and are two exact match
pairs, we know
. Since and are two exact match
pairs, we know
Obviously, the gap lengthes between them are
Introduce 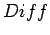 , number of base differences resulting from
extension, as
, number of base differences resulting from
extension, as
where
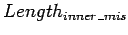 is the number of different(mismatches
and indels) bases from global alignment of
is the number of different(mismatches
and indels) bases from global alignment of
![$s[i_{e}+1\ldots
m_{b}-1]$](img33.gif) and
and
![$s[j_{e}+1\ldots n_{b}-1]$](img34.gif) . The similarity of merged
loci is then:
. The similarity of merged
loci is then:
When LTR_FINDER decides whether two neighboring pairs should be
merged, it first calculates , make sure that it does not
exceed the value of extension max gap, then calculates 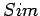 .
If
.
If
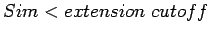 , pair extension will stop here,
, pair extension will stop here,
![$P_1\{s[i_b\ldots i_e], s[j_b\ldots j_e]\}$](img38.gif) will be reported as a candidate for LTR element; If
will be reported as a candidate for LTR element; If
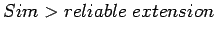 , new pair and inter-pair
regions will be linked to the previous one to construct a longer new
pair
, new pair and inter-pair
regions will be linked to the previous one to construct a longer new
pair
![$P\{s[i_b\ldots m_e], s[j_b\ldots n_e]\}$](img40.gif) , and LTR_FINDER continues to find next neighboring pairs; if
, and LTR_FINDER continues to find next neighboring pairs; if
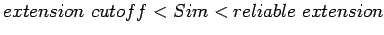 , it means we are not sure whether continue to extend or stop. So LTR_FINDER first report a LTR element candidate
while at the same time, the extension process will continue.
, it means we are not sure whether continue to extend or stop. So LTR_FINDER first report a LTR element candidate
while at the same time, the extension process will continue.
Running time of LTR_FINDER is very sensitive to this parameter. The
program only selects pairs that are longer than this value to do
further processes from all exact match pairs detected. If a very
small value is given, LTR_FINDER will spend much time on randomly
matched short sequences. We use P-value to estimate proper value of
-p, that is, the probability of exact match of length longer than
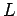 occurs if 2 sequences are drown randomly. In Waterman
1989(, ), under independent letter model, using
asymptotic extreme value distribution, P-value were worked out
analytically. Now if one assign the P-value, length can be deduced.
From experiments, 20 is appropriate in most situation, and we
suggest not using of value less than 15.
occurs if 2 sequences are drown randomly. In Waterman
1989(, ), under independent letter model, using
asymptotic extreme value distribution, P-value were worked out
analytically. Now if one assign the P-value, length can be deduced.
From experiments, 20 is appropriate in most situation, and we
suggest not using of value less than 15.
When aligning tRNA 3' tail 18nt string to the inter-LTR sequence, if
LTR_FINDER finds an alignment that match length exceeds this
threshold, it will report this region as a putative primer binding
site.
To predict primer binding site, we need tRNA sequences, especially
the 3' end 18nt of each sequences. LTR_FINDER can load tRNA of
different species. A good tRNA set can be found at Genomic tRNA
Database (http://lowelab.ucsc.edu/GtRNAdb/). Our database was download from Genomic tRNA Database on 2007-07-17. Here is an example of required format:
>Athal-chr4.trna25-AlaAGC (13454563-13454635)
GGGGATGTAGCTCAGATGGTAGAGCGCTCGCTTAGCATGCGAGAGGCACGGGGATCGATA
CCCCGCATCTCCA
LTR_FINDER uses the string after the last minus sign in sequence
identifier field as the tRNA type name. In this example, `AlaAGC'.
This parameter is a directory name. LTR_FINDER can predict protein
domains by calling ps_scan, which can be obtained from
ExPASy-PROSITE (http://www.expasy.org/prosite/). User should place
data file `prosite.dat' and ps_scan in this directory. If this
parameter is enabled, LTR_FINDER will call them and report these
protein domains if they are detected.
This is the threshold for LTR retrotransposon score. Models that
have higher scores are output.
LTR_FINDER calculates sharpness for both 5' and 3' extremities of
putative elements. Both of them must be greater than the lower
threshold and one greater than the higher threshold.
This parameter controls the output format:
| Value |
Format |
| 0 |
Full output |
| 1 |
Summary output |
| 2 |
Table output(not available on web) |
LTR_FINDER can output alignment details of LTR boundaries (left to
120bp and right to 80bp relative to boundaries). Users are allowed
to assign the output length by setting this parameter. The whole 200
bp alignment is output when it 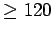 .
.
This parameter is ID of one sequence in a multi-fasta file. When
enabled, only sequences with this ID will be processed.
The parameter controls output by sequence tag status. It is a binary
string of 11 bits. From left to right, bits denote the status of
following signals: TG in 5' end of 5'LTR, CA in 3' end of 5'LTR, TG
in 5' end of 3'LTR, CA in 3' end of 3'LTR, TSR, PBS, PPT, RT,
IN(core), IN(c-term) and RH. 1 means reported models should
containing the signal and 0 ignoring searching it. when used, the
program will only report models whose status of sequence tags match
it.
With this parameter, LTR_FINDER will try to mask highly repeated regions defined by: the same exact match pair repeat 14 more times within 3000bp. By using this parameter, LTR_FINDER can perform much more quickly on sequences which have centriole or telomere region.
We thank Heng Li for his linear-space pairwise alignment library and Xiaoli Shi for providing rice tRNA sequences. The authors are also grateful to colleagues who helped us testing the web server.
-
Flavell, R. B. (1986).
- Repetitive dna and chromosome evolution in plants.
Philos Trans R Soc Lond B Biol Sci, 312(1154),
227-242.
-
Gao, L., McCarthy, E. M., Ganko, E. W., and McDonald, J. F. (2004).
- Evolutionary history of oryza sativa ltr retrotransposons: a
preliminary survey of the rice genome sequences.
BMC Genomics, 5(1), 18.
-
Gao, X., Havecker, E. R., Baranov, P. V., Atkins, J. F., and Voytas, D. F.
(2003).
- Translational recoding signals between gag and pol in diverse ltr
retrotransposons.
RNA, 9(12), 1422-1430.
-
Jordan, I. K. and McDonald, J. F. (1998).
- Evidence for the role of recombination in the regulatory evolution of
saccharomyces cerevisiae ty elements.
J Mol Evol, 47(1), 14-20.
-
Kalyanaraman, A. and Aluru, S. (2006).
- Efficient algorithms and software for detection of full-length ltr
retrotransposons.
J Bioinform Comput Biol, 4(2), 197-216.
-
Kim, J. M., Vanguri, S., Boeke, J. D., Gabriel, A., and Voytas, D. F. (1998).
- Transposable elements and genome organization: a comprehensive survey
of retrotransposons revealed by the complete saccharomyces cerevisiae genome
sequence.
Genome Res, 8(5), 464-478.
-
Ma, J., Devos, K. M., and Bennetzen, J. L. (2004).
- Analyses of ltr-retrotransposon structures reveal recent and rapid
genomic dna loss in rice.
Genome Res, 14(5), 860-869.
-
McCarthy, E. M. and McDonald, J. F. (2003).
- Ltr_struc: a novel search and identification program for ltr
retrotransposons.
Bioinformatics, 19(3), 362-367.
-
McCarthy, E. M. and McDonald, J. F. (2004).
- Long terminal repeat retrotransposons of mus musculus.
Genome Biol, 5(3), R14.
-
McCarthy, E. M., Liu, J., Lizhi, G., and McDonald, J. F. (2002).
- Long terminal repeat retrotransposons of oryza sativa.
Genome Biol, 3(10), RESEARCH0053.
-
McDonald, J. F. (1993).
- Evolution and consequences of transposable elements.
Curr Opin Genet Dev, 3(6), 855-864.
-
McDonald, J. F., Matyunina, L. V., Wilson, S., Jordan, I. K., Bowen, N. J., and
Miller, W. J. (1997).
- Ltr retrotransposons and the evolution of eukaryotic enhancers.
Genetica, 100(1-3), 3-13.
-
SanMiguel, P., Tikhonov, A., Jin, Y. K., Motchoulskaia, N., Zakharov, D.,
Melake-Berhan, A., Springer, P. S., Edwards, K. J., Lee, M., Avramova, Z.,
and Bennetzen, J. L. (1996).
- Nested retrotransposons in the intergenic regions of the maize
genome.
Science, 274(5288), 765-768.
-
Xiong, Y. and Eickbush, T. H. (1990).
- Origin and evolution of retroelements based upon their reverse
transcriptase sequences.
EMBO J, 9(10), 3353-3362.
-
Yoder, J. A., Walsh, C. P., and Bestor, T. H. (1997).
- Cytosine methylation and the ecology of intragenomic parasites.
Trends Genet, 13(8), 335-340.
-
Zhang, X. and Wessler, S. R. (2004).
- Genome-wide comparative analysis of the transposable elements in the
related species arabidopsis thaliana and brassica oleracea.
Proc Natl Acad Sci U S A, 101(15), 5589-5594.
LTR_FINDER USER MANUAL
version 1.0.2
This document was generated using the
LaTeX2HTML translator Version 2008 (1.71)
Copyright © 1993, 1994, 1995, 1996,
Nikos Drakos,
Computer Based Learning Unit, University of Leeds.
Copyright © 1997, 1998, 1999,
Ross Moore,
Mathematics Department, Macquarie University, Sydney.
The command line arguments were:
latex2html -up_url http://tlife.fudan.edu.cn/ltr_finder -up_title 'LTR_FINDER Home Page' -transparent -antialias_text -antialias -image_type gif -local_icons -split 0 help
The translation was initiated by on 2009-04-09
Up: LTR_FINDER Home Page
2009-04-09